Every year, thousands of children in India are born with thalassemia major, a serious inherited blood disorder that affects the body’s ability to produce healthy hemoglobin. For many families, the diagnosis brings lifelong dependence on blood transfusions, emotional stress, and financial uncertainty. Yet advances in modern hematology and stem cell transplantation have dramatically changed the outlook for patients living with this condition.
India has emerged as one of the leading destinations for comprehensive Thalassemia Treatment in India. The country now offers advanced diagnostic facilities, experienced hematologists, internationally accredited transplant units, and affordable treatment pathways compared with many Western nations. From long-term supportive therapy to potentially curative bone marrow transplantation, patients today have access to a wide range of medical options that can significantly improve both survival and quality of life.
What Is Thalassemia?
Thalassemia is an inherited blood disorder that affects the body’s ability to produce normal hemoglobin, the oxygen-carrying protein found in red blood cells. As a result, red blood cells become fragile and break down more quickly, causing anemia and reduced oxygen supply to the body’s tissues.
The condition is passed genetically from parents to children through an autosomal recessive pattern of inheritance. Thalassemia can vary in severity, ranging from mild forms with few or no symptoms to severe cases that require ongoing medical care, including regular blood transfusions and specialized treatment.
Types of Thalassemia
A. Thalassemia Major
Also called Beta-Thalassemia Major or Cooley’s Anemia, this is the most severe form of the disease. Children with Thalassemia Major typically present by the age of two with profound anemia, failure to thrive, pale skin, and progressive enlargement of the spleen and liver. Without treatment, the condition is fatal.
- Symptoms: Severe fatigue, bone deformities (particularly of the face and skull), growth retardation, jaundice, enlarged spleen (splenomegaly), and frequent infections.
- Treatment dependency: Patients require blood transfusions every 2–4 weeks for life. Over time, each transfusion deposits excess iron in vital organs, making iron chelation therapy essential alongside transfusions.
- Long-term risks: Iron overload leading to cardiac failure, liver cirrhosis, and endocrine complications (diabetes, hypothyroidism, delayed puberty). The only curative treatment is a bone marrow transplant.
B. Thalassemia Intermedia
This form sits between the minor and major classifications. Patients with Thalassemia Intermedia produce some functional hemoglobin but not enough to sustain normal health without periodic medical intervention.
- Symptoms: Moderate anemia, bone changes, mild to moderate splenomegaly, occasional jaundice, and exercise intolerance.
- Treatment dependency: Transfusions are intermittent rather than regular — often triggered by infections or physiological stress. Many patients manage for years without them, but complications accumulate silently.
- Long-term risks: Pulmonary hypertension, leg ulcers, thrombosis, and progressive iron accumulation even without transfusions. Monitoring and timely escalation to transplant evaluation are critical.
C. Thalassemia Minor
Individuals with Thalassemia Minor inherit only one defective gene and are clinical carriers. They typically live normal lives with at most mild anemia.
- Symptoms: Usually none, or subtle fatigue that is often dismissed as lifestyle-related.
- Why it matters: Two carriers have a 25% chance with each pregnancy of having a child with Thalassemia Major. Genetic counseling and prenatal testing are non-negotiable for carrier couples. Simple blood tests like HbA2 estimation (part of a complete hemoglobin electrophoresis) can identify carrier status accurately.
Symptoms of Thalassemia
Whether you are evaluating a child or an adult, recognizing the signs early leads to faster diagnosis and better outcomes. Common symptoms across the spectrum include:
- Persistent fatigue and weakness that does not resolve with rest or nutritional changes is often the first sign.
- This is accompanied by pallor — a noticeably pale or yellowish tone to the skin, gums, and inner eyelids. In children, delayed growth and development is common, as chronic anemia deprives growing tissues of oxygen.
- As the disease progresses, bone deformities — especially a characteristic protruding forehead and prominent cheekbones caused by expanded bone marrow — become visible.
- The spleen works overtime to clear damaged red blood cells, leading to splenomegaly (enlarged spleen), which can cause abdominal discomfort and a sense of fullness. Frequent infections, dark urine, and jaundice round out the clinical picture in more advanced cases.
If your child is showing two or more of these signs, consult a pediatric hematologist without delay.
Causes and Genetic Inheritance
Thalassemia is caused by mutations in the genes responsible for hemoglobin chain production — specifically the alpha- and beta-globin genes on chromosomes 16 and 11, respectively.
The disease follows autosomal recessive inheritance; both parents must carry at least one mutated copy for a child to inherit the disorder. When both parents are carriers, each pregnancy carries:
- A 25% chance of a child with Thalassemia Major
- A 50% chance of a child who is a carrier (Thalassemia Minor)
- A 25% chance of an entirely unaffected child
Prenatal diagnosis — through chorionic villus sampling (CVS) at 10–12 weeks or amniocentesis at 14–16 weeks — can identify affected fetuses early, giving families the chance to make informed decisions. Preimplantation Genetic Diagnosis (PGD) combined with IVF is now available in India for high-risk couples, allowing only unaffected embryos to be transferred.
Family history, consanguineous marriages, and populations from the Indian subcontinent, Mediterranean, Southeast Asia, and the Middle East carry higher-than-average carrier rates. Screening entire families when one member is diagnosed is strongly recommended.
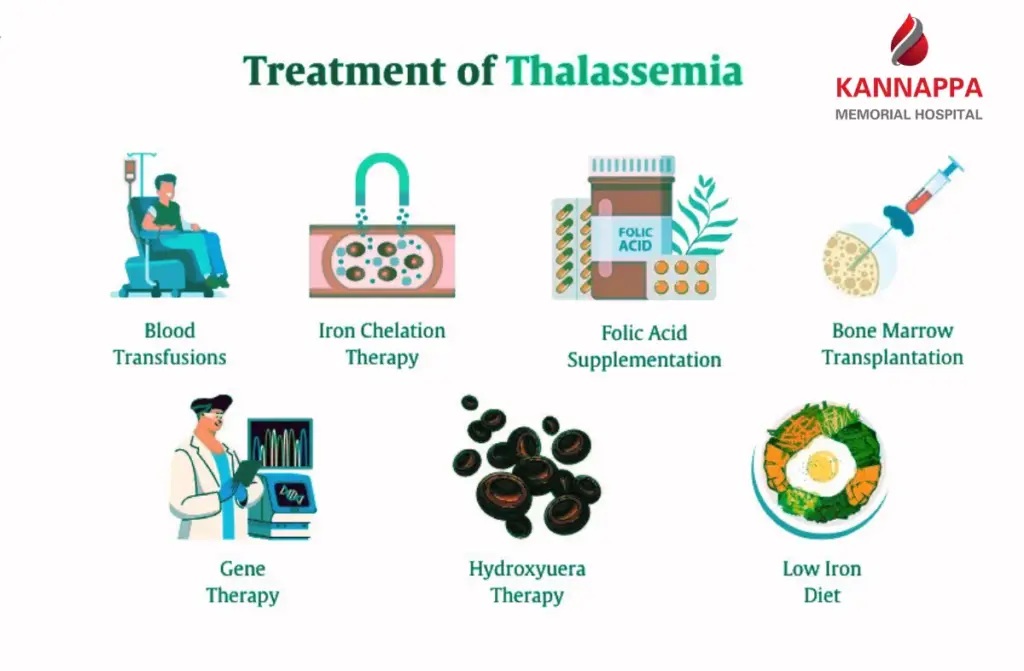
Thalassemia Treatment Options
Blood Transfusion Therapy
- Regular blood transfusions are the backbone of supportive thalassemia treatment in India. Packed red blood cell (PRBC) transfusions, typically administered every 2–4 weeks, maintain hemoglobin levels above 9–10 g/dL and suppress the body’s ineffective bone marrow activity.
- Each session lasts 3–6 hours. While blood transfusions are essential for survival, repeated transfusions over many years can lead to iron overload, where excess iron builds up in the body and damages vital organs.
- Since the body has no natural mechanism to excrete excess iron from transfused cells, it accumulates in the liver, heart, and endocrine organs.
Iron Chelation Therapy
- Iron chelation is the medical management of iron overload caused by repeated transfusions. Chelating agents attach to excess iron in the body and help remove it through urine and stool. Iron chelation therapy in India costs between INR 10,000 to 50,000 per month, depending on the drug used. The three main agents are Deferoxamine (injectable), Deferiprone (oral), and Deferasirox (oral, once daily). Regular monitoring through serum ferritin levels and MRI-based liver iron concentration (LIC) assessments guides dosing. Without adequate chelation, iron overload remains the leading cause of cardiac death in thalassemia patients.
Bone Marrow Transplant
A bone marrow transplant , also known as allogenic hematopoietic stem cell transplantation (HSCT), is currently the only proven cure for Thalassemia Major.
- Donor matching: The ideal donor is an HLA (Human Leukocyte Antigen)-matched sibling. When no matched sibling is available, a matched unrelated donor (MUD) sourced from national or international donor registries is used. Haploidentical transplants (half-matched, typically a parent) are increasingly successful with modern protocols.
- Eligibility: The best transplant outcomes are usually seen in younger children, especially those under 7–8 years of age, who have minimal organ damage and well-controlled iron levels. However, older patients and individuals with existing organ complications may also undergo transplantation using specialized treatment protocols tailored to their condition.
- Success rates: Thalassemia stem cell transplant in India achieves 80–90% success rates in well-matched sibling donor cases at experienced centers. Outcomes in unrelated donor transplants have improved significantly with better HLA typing and infection control.
Stem Cell Transplant
- While “bone marrow transplant” is the common term, stem cells can be harvested from three sources: the bone marrow itself, peripheral blood (after G-CSF stimulation), and umbilical cord blood.
- Peripheral blood stem cell transplants (PBSCT) offer faster engraftment but carry a higher risk of chronic GVHD. Cord blood transplants are an emerging option when no adult donor is available, particularly in younger, smaller children.
Hormonal and Supportive Therapy
- Long-term thalassemia and iron overload can affect hormone production and growth. Some patients may develop conditions such as thyroid problems, diabetes, delayed growth, or delayed puberty.
- Doctors may recommend hormone treatment, nutritional support, and regular health checkups to manage these issues and improve quality of life.
Genetic Screening and Counseling
- Preventing thalassemia is very important. Genetic screening can identify thalassemia carriers before marriage or pregnancy, while prenatal testing and genetic counseling help families understand the chances of passing the condition to their children.
- Many screening and awareness programs are available across India to help reduce the number of children born with severe thalassemia.
Risks and Complications
- Thalassemia and its treatments can sometimes lead to serious complications, especially with long-term transfusions and bone marrow transplant procedures.
- One of the biggest risks is iron overload caused by repeated blood transfusions. Extra iron can build up in the liver, heart, and other organs, leading to problems such as liver damage, heart disease, and hormonal disorders if not properly treated with iron chelation therapy.
- After a bone marrow transplant, infections are a major concern because the immune system becomes weak during treatment. Patients require close monitoring and protective care to reduce infection risk.
- Another possible complication is Graft-Versus-Host Disease (GVHD), where the donor’s immune cells attack the patient’s body tissues. Some patients may develop mild to severe GVHD affecting the skin, liver, or digestive system. Chronic GVHD can also occur in a smaller number of patients.
- In rare cases, the transplant may fail if the donor cells do not grow properly inside the patient’s bone marrow. This is called graft failure and may require additional treatment or another transplant.
Cost of Thalassemia Treatment in India
Supportive Care Costs
Blood transfusions cost between INR 1,000 to 3,000 per session, with most patients requiring 12–26 sessions per year. Iron chelation adds INR 1.2–6 lakhs annually. Diagnostics, outpatient visits, and complications push annual supportive care costs significantly higher over time — making curative treatment the economically rational long-term choice.
Bone Marrow Transplant Cost
Thalassemia treatment cost in India ranges from INR 12.5 lakhs to INR 33.5 lakhs (approximately $15,000 to $40,000), depending on transplant type and donor source. Unrelated donor transplants, which involve HLA typing and donor registry procurement, can range from INR 15 lakhs to INR 25 lakhs and higher for complex cases. Packages typically cover hospital stay, conditioning chemotherapy, the transplant procedure, and initial recovery.
Financial Support
The Government of India’s Thalassemia Bal Sewa Yojana provides financial assistance of up to INR 10 lakhs per patient for eligible families with an annual income below INR 8 lakhs. Several hospitals offer EMI-based payment plans. Many insurance providers cover hospitalization costs for bone marrow transplant. International patients are advised to request detailed cost packages including accommodation and follow-up care estimates before travel.
Conclusion
Thalassemia is a lifelong challenge — but it is no longer a life sentence. With the right diagnosis, expert management, and access to curative bone marrow transplant, children with Thalassemia Major can grow up to lead healthy, transfusion-free lives. Thalassemia treatment in India today represents a convergence of clinical expertise, modern infrastructure, and genuine affordability — making world-class outcomes accessible to families across India and internationally.
The earlier the intervention, the better the outcome. Whether you are newly diagnosed, managing a child on transfusions, or actively exploring transplant options, do not delay consultation with a specialist. The path to cure exists — and it begins with informed action.
Read also Bone Marrow Transplant Success Rate in India.